Noise and Gene Expression
The ability to construct synthetic gene networks enables detailed experimental investigations of deliberately simplified systems that can be compared to quantitative models. If simple, well-characterized modules can be coupled together into more complex networks whose behavior is then predictable from that of the individual components, we may begin to build an understanding of cellular regulatory processes from the bottom up.
In this work an engineered promoter that allowed the simultaneous repression and activation of gene expression in Escherichia coli was constructed and used to study synthetic gene networks under increasingly complex conditions: unregulated, repressed, and simultaneously activated and repressed.
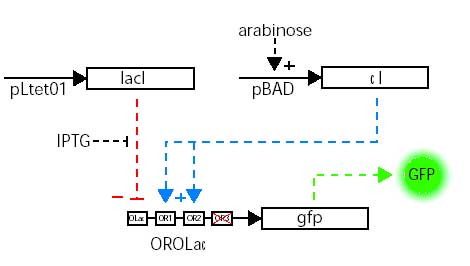
A stochastic model that quantitatively captured the means and distributions of the expression from the engineered promoter of this modular system was constructed and shown to accurately predict the in vivo behavior of an expanded network that included positive feedback. The model also revealed the counterintuitive prediction that noise in protein expression levels can increase upon arrest of cell division, which was confirmed experimentally. (In collaboration with James Collins’s laboratory)
Guido NJ, Wang X, Adalsteinsson D, McMillen D, Hasty J, Cantor CR, Elston TC, Collins JJ. A bottom-up approach to gene regulation. Nature. 2006 Feb 16; 439(7078):856-60
(Pubmed | Journal)
Fluorescent Proteins as Quantitative Tools
 Fluorescent proteins are often used as reporters of transcriptional activity. Here we present a mathematical characterization of a novel fluorescent reporter that was recently engineered to have a short half-life (~12 min). The advantage of this destabilized protein is that it can track the transient transcriptional response often exhibited by signaling pathways. Our mathematical model takes into account the maturation time and half-life of the fluorescent protein. We demonstrate that our characterization allows transient transcript profiles to be inferred from fluorescence data. We also investigate a stochastic version of the model. Our analysis reveals that fluorescence measurements can both underestimate and overestimate fluctuations in protein levels that arise from the stochastic nature of biochemical reactions.
Fluorescent proteins are often used as reporters of transcriptional activity. Here we present a mathematical characterization of a novel fluorescent reporter that was recently engineered to have a short half-life (~12 min). The advantage of this destabilized protein is that it can track the transient transcriptional response often exhibited by signaling pathways. Our mathematical model takes into account the maturation time and half-life of the fluorescent protein. We demonstrate that our characterization allows transient transcript profiles to be inferred from fluorescence data. We also investigate a stochastic version of the model. Our analysis reveals that fluorescence measurements can both underestimate and overestimate fluctuations in protein levels that arise from the stochastic nature of biochemical reactions.
Wang X, Errede B, Elston TC. Mathematical analysis and quantification of fluorescent proteins as transcriptional reporters. Biophys J. 2008 Mar 15; 94(6):2017-26. (Pubmed | Journal)
The Role of a Tumor Suppressor Gene p16INK4a in Aging
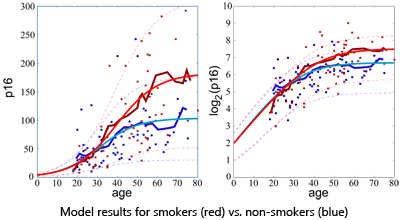 Recent work has shown that expression of the p16INK4a tumor suppressor increases with chronological age. Expression is accelerated by geron-togenic behaviors such as tobacco use and physical inactivity, and is also influenced by allelic genotype of a polymorphic single nucleotide poly- morphism (SNP) rs10757278 that is physically linked with the p16INK4a ORF. To understand the relationship between p16INK4a expression, chronologic age, subject characteristics and host genetics, we developed a mathematical model that links p16INK4a expression with aging. Using an annotated dataset of 170 healthy adults for whom p16INK4a expression and subject genotypes were known, we studied two alternative stochastic models that relate p16INK4a expression to age, smoking, exercise and rs10757278 genotype. Levels of p16INK4a increased exponentially and then saturated at later chronologic ages. The model, which best fit the data, suggests saturation occurs because of p16INK4a-dependent attrition of subjects at older chronologic ages, presumably due to death or chronic illness. An important feature of our model is that factors that contribute to death in a non p16INK4a-dependent manner do not affect our analysis. Interestingly, tobacco-related increases in p16INK4a expression are predicted to arise from a decrease in the rate of p16INK4a-dependent death. This analysis is most consistent with the model that p16INK4a expression monotonically increases with age, and higher expression is associated with increased subject attrition.
Recent work has shown that expression of the p16INK4a tumor suppressor increases with chronological age. Expression is accelerated by geron-togenic behaviors such as tobacco use and physical inactivity, and is also influenced by allelic genotype of a polymorphic single nucleotide poly- morphism (SNP) rs10757278 that is physically linked with the p16INK4a ORF. To understand the relationship between p16INK4a expression, chronologic age, subject characteristics and host genetics, we developed a mathematical model that links p16INK4a expression with aging. Using an annotated dataset of 170 healthy adults for whom p16INK4a expression and subject genotypes were known, we studied two alternative stochastic models that relate p16INK4a expression to age, smoking, exercise and rs10757278 genotype. Levels of p16INK4a increased exponentially and then saturated at later chronologic ages. The model, which best fit the data, suggests saturation occurs because of p16INK4a-dependent attrition of subjects at older chronologic ages, presumably due to death or chronic illness. An important feature of our model is that factors that contribute to death in a non p16INK4a-dependent manner do not affect our analysis. Interestingly, tobacco-related increases in p16INK4a expression are predicted to arise from a decrease in the rate of p16INK4a-dependent death. This analysis is most consistent with the model that p16INK4a expression monotonically increases with age, and higher expression is associated with increased subject attrition.
Tsygankov D, Liu Y, Sanoff HK, Sharpless NE, Elston TC. A quantitative model for age-dependent expression of the p16INK4a tumor suppressor. Proc Natl Acad Sci U S A. 2009 Sep 29; 106(39): 16562-16567.
(Pubmed | Journal)
Further Publications
Guido NJ, Lee P, Wang X, Elston TC, Collins JJ. A pathway and genetic factors contributing to elevated gene expression noise in stationary phase. Biophys J. 2007 Dec 1; 93(11):L55-7
(Pubmed | Journal)
Erban R, Frewen TA, Wang X, Elston TC, Coifman R, Nadler B, Kevrekidis IG. Variable-free exploration of stochastic models: a gene regulatory network example. J Chem Phys. 2007 Apr 21; 126(15):155103
(Pubmed | Journal)
Wang X, Hao N, Dohlman HG, Elston TC. Bistability, stochasticity, and oscillations in the mitogen-activated protein kinase cascade. Biophys J. 2006 Mar 15; 90(6):1961-78
(Pubmed | Journal)
Erban R, Kevrekidis IG, Adalsteinsson D, Elston TC. Gene regulatory networks: a coarse-grained, equation-free approach to multiscale computation. J Chem Phys. 2006 Feb 28; 124(8):084106
(Pubmed | Journal)
Kaern M, Elston TC, Blake WJ, Collins JJ. Stochasticity in gene expression: from theories to phenotypes.Nat Rev Genet. 2005 Jun; 6(6):451-64.
(Pubmed | Journal)
Pirone JR, Elston TC. Fluctuations in transcription factor binding can explain the graded and binary responses observed in inducible gene expression. J Theor Biol. 2004 Jan 7; 226(1):111-21
(Pubmed | Journal)
Adalsteinsson D, McMillen D, Elston TC. Biochemical Network Stochastic Simulator (BioNetS): software for stochastic modeling of biochemical networks. BMC Bioinformatics. 2004 Mar 8; 5:24
(Pubmed | Journal)